cmuts core
Purpose
cmuts core performs the main job of the cmuts pipeline, which is determining the location and type of mutations, insertions, and deletions in a collection of aligned reads.
Usage
All modes of cmuts core require two inputs to run:
- Reference sequences, stored in a FASTA file,
- Aligned reads, stored in one or more SAM/BAM/CRAM files
The basic syntax is
It is recommended to use default settings alongside the --no-insertions flag when processing chemical probing data.
Warning
The alignments must be sorted before passing to cmuts. If you are generating them via cmuts align, then they will automatically be sorted for you.
Modification Counting
Most uses of cmuts core (without any of the flags specified later) will output an HDF5 file with a dataset of shape \(n \times l \times 4 \times 7\). The former two dimensions specify the reference sequence and residue respectively, and the latter two specify the type of modification seen in accordance with the following array:
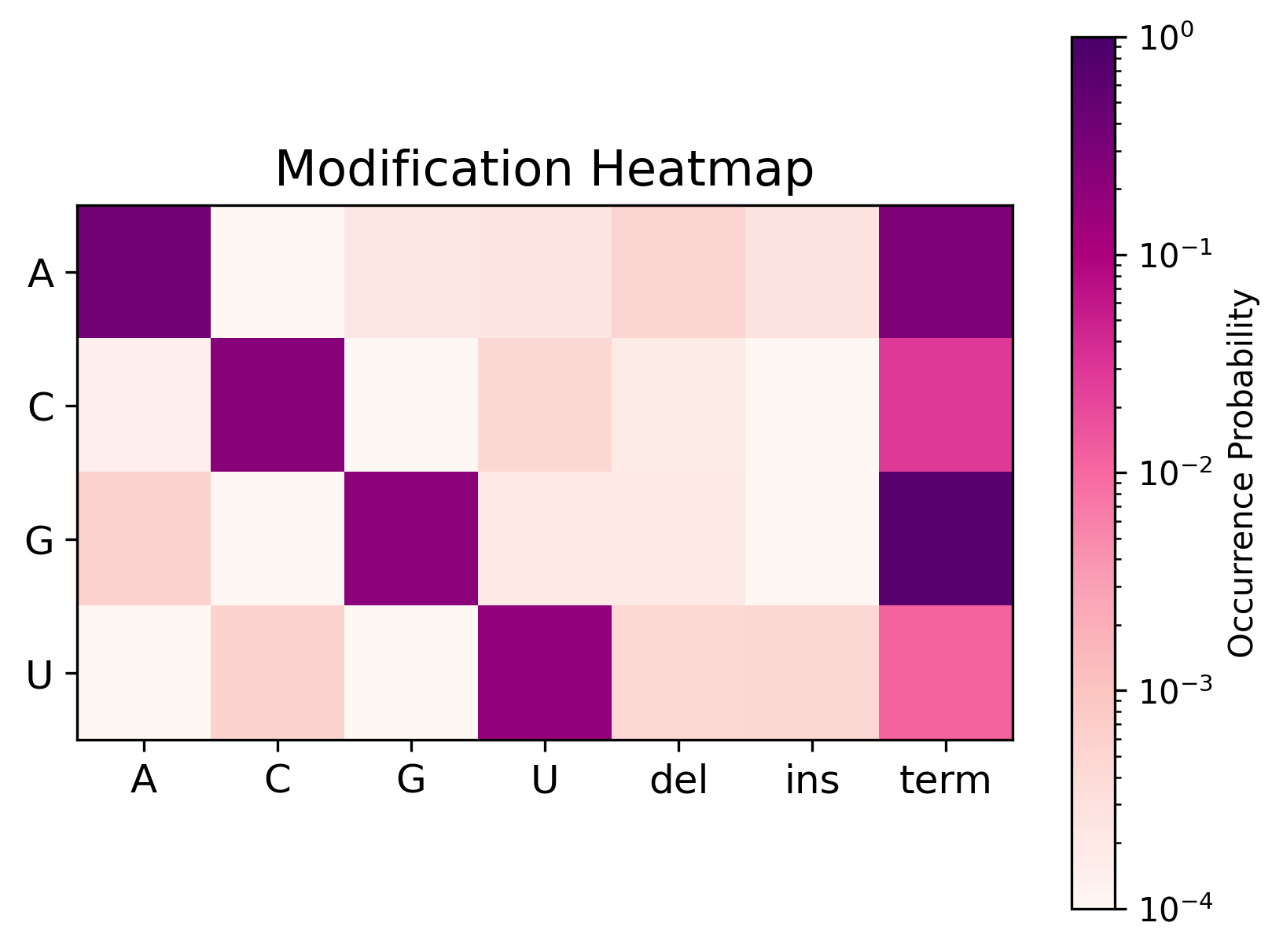
The diagonal corresponds to matches, whereas the off-diagonal corresponds to mismatches. The final three columns correspond to deletions, insertions, and termination events respectively.
The name of the dataset is counts-1d, and the group to which it belongs corresponds to the name of the input file. For example, the command
will create an HDF5 file $OUTPUT with the structure
and both IN1 and IN2 are datasets as described above.
Pairwise Modification Counting
The --pairwise flag instructs cmuts core to count pairs of modifications alongside the one-dimensional data. In addition to the above output, the HDF5 file will contain a second dataset counts-2d of shape \(n \times l \times l \times 2 \times 2\). The first dimension specifies the reference sequence, the next two specifies each pair of bases in that sequence, and the final two specify the four entries of the joint Bernoulli distribution
Again for an example, the command
will create an HDF5 file $OUTPUT with the structure
Command Line Options
Input/Output
files : The input SAM/BAM/CRAM files.
-o, --output : The output HDF5 file. (required)
-f, --fasta : The reference FASTA file. (required)
--overwrite : Overwrite an existing HDF5 file.
--rebuild : Rebuild all index files.
-c, --compression : Compression level of the HDF5 output (0-9). (default: 3)
--print-every : How often (in seconds) to print statistics. (default: 0.01)
-v, --verbose : Enable verbose (debug) logging to .cmuts.log file.
Mode
--pairwise : Compute pairwise modification counts.
--tokenize : Tokenize the reference sequences.
--token-map : Comma-separated non-negative integer tokens for A,C,G,U in order, used by --tokenize (e.g. "4,5,6,7"). T and U share the fourth value. Defaults to 0,1,2,3.
Filtering
--min-mapq : Mapping quality threshold for alignment processing. (default: 10)
--min-phred : PHRED score threshold for base processing. (default: 10)
--min-length : Skip reads shorter than this length. (default: 2)
--max-length : Skip reads longer than this length. (default: 1024)
--max-hamming : The maximum number of mismatches, insertions, and deletions in a processed read. (default: 1024)
--secondary : Consider secondary alignments for processing as well.
--downsample : Process at most this many quality-passing reads per reference (caps usable depth and avoids over-processing). Reads failing the quality filters do not count toward the limit.
--ignore-bases : Do not count mismatches or deletions occurring at these bases. Pass as a single string.
Processing
--max-indel-length : The longest indels to consider. (default: 10)
--quality-window : Check the quality of each base in a window of this size around each base. (default: 2)
--collapse : Collapse modifications within this distance of each other in a given read. (default: 2)
Performance
--chunk-size : The number of references to process at a time per thread. (default: 128)
Mutation type filters
--no-mismatches : Do not count mismatches as modifications.
--no-insertions : Do not count insertions as modifications.
--no-deletions : Do not count deletions as modifications.
Strand options
--no-reverse : Ignore reverse-complemented reads.
--only-reverse : Use only reverse-complemented reads.
Ambiguous Deletions
--uniform-spread : Uniformly spread out ambiguous deletions.
--no-spread : Do not spread ambiguous deletions.
--disable-ambiguous : Disable the ambiguous deletion detection algorithm, relying on the deletion provided by the alignment.
Quality filtering
--no-match-filter : Do not filter matches based on their PHRED base score.
--no-insertion-filter : Do not filter insertions based on their PHRED base score.
--no-deletion-filter : Do not filter deletions based on their PHRED base score.